Project Summary
The evolution of a cancer system consisting of cancer clones and normal cells is a complex and dynamic process with multiple interacting factors including clonal expansion, somatic mutation, and sequential selection. As a typical example, in patients with chronic lymphocytic leukemia (CLL), a monoclonal population of transformed B cells expands to dominate the B cell population in the peripheral blood and bone marrow. This expansion of transformed B cells suggests that they might evolve through processes distinct from those of normal B cells. Recent advances in next generation sequencing enable the high-throughput identification and tracking of individual B cell clones through sequencing of the V-D-J junction segments of the immunoglobulin heavy chain (IGH). Here, we developed a statistical approach to modeling cellular evolution of the immune repertoire. Adapting the infinitely many alleles model from population genetics, we derived the Ewens sampling test (EST) to distinguish immune repertoires of CLL patients with imminent relapse from healthy controls and patients in sustained remission. Extensive simulations showed that EST is sensitive in detecting cancer-related derangements of the IGH repertoire. In addition, we developed two potentially useful parameters: the rate at which donor’s B cell clones enter the circulation and the average time to regenerate a transplanted immune repertoire, both of which provide additional information about the dynamics of immune reconstitution in these patients. We intend to apply our models and statistics in other large-scale clinical studies, including T-ALL study via collaboration with AdaptiveTCR and the large-scale CLL study through collaboration with Sequenta.
Significance
The immune repertoire of an individual is the collection of antigen receptors of B or T cells in circulation at a given time. The dynamics of the immune repertoire is dictated by various diseases, and serve as an important indicator of the competence of the adaptive immune system. Monitoring the adaptive immune system could help classify and diagnose human diseases.
Next-generation sequencing (NGS) enables the comprehensive examination of immune repertoire. However, statistical and computational approaches remain to be developed to characterize and detect the changes of the state of the immune repertoire.
Approach
Sequencing the immune repertoire can provide important insights into the nature of diseases. CLL is B cell malignancies, which are clonal cells sharing antigen receptors with an identical genomic rearrangement of V-D-J gene segments. Previously, we used 454 technology to sequence B cell immunoglobulin heavy chain receptors in donors and CLL patients at diagnosis and at multiple time points after allo-HCT .

We model the evolution of immune repertoire with the Moran infinitely many alleles model assuming a roughly constant population size. We made a simplifying assumption that after novel B cells acquire VDJ rearranged antigen receptors and mature in bone marrow or lymph nodes, they enter the circulation and proliferate independently and spontaneously in the absence of antigen stimulation.

The Ewens sampling distribution, which is derived from the infinite many alleles model, can describe this V-J combination frequency pattern of a snapshot of the healthy immune repertoire.
We first performed simulation to demonstrate our model may be a reasonable fit to the data.

Next we devised the Ewens sampling test (EST) and two estimators from the infinite many alleles model. We showed the sensitivity of EST and the immigration rate θ via simulation.
We then applied EST to the sequencing data of immune repertoires of patient samples. Both donor samples and samples in remission have pvalue 1 while cancer samples have significant pvalues.

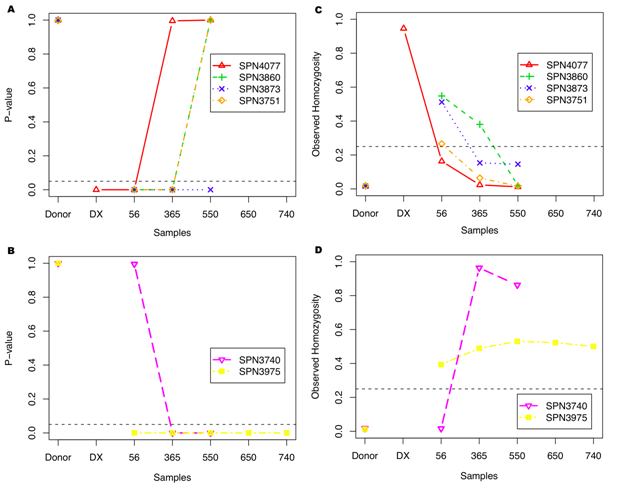

We estimated the immigration rate θ of novel B cells to dynamically monitor the evolutionary status of immune repertoire. Figures above show rate θ increases as patients recover while θ decreases as CLL relapses. This estimator reveals the evolutionary dynamics of immune repertoire.
Furthermore, we estimated the average time to re-generate the immune repertoire observed at a given time based on our stochastic model after removing any cancer clones and subclones. It is of clinical interest, as it measures the expansion or shrinkage rates of the donor’s immune repertoire in the recipient’s circulation, which can be used to predict the patient recovery status after treatments.
Accomplishments
The infinite many alleles model well describes the normal pattern of immune repertoire. We applied the Ewens sampling test derived from this model to detect CLL. We also developed two estimators of clinical importance, the immigration rate of novel B cells and the average time to reconstruct the healthy repertoire after transplantation.
Future Objectives
We intend to apply our models and statistics in other large-scale clinical studies, including T-ALL study via collaboration with AdaptiveTCR and the large-scale CLL study through collaboration with Sequenta.
Acknowledgements
We thank Dr. Hua Chen, Dr. Junhee Seok and Dr. Aaron Logan for helpful discussion. This work was supported by Stanford Genome Technology Center Grant (NIH P01-HG000205).
Personnel
Hong Gao1, Chunlin Wang1, Aaron C. Logan2, Carlos D. Bustamante3, Michael Mindrinos1, David Miklos2, Marcus W. Feldman4, Ronald W. Davis1 and Wenzhong Xiao1,5
1 Stanford Genome Technology Center and Biochemistry Department, Stanford University, Stanford, CA.;
2 Division of Blood and Marrow Transplantation, Department of Medicine, Stanford University, Stanford, CA;
3 Department of Genetics, Stanford University, CA;
4 Department of Biology, Stanford University, CA;
5 Massachusetts General Hospital, Harvard Medical School, Shriners Hospital for Children, Boston, MA.